Hospital Universitario La Moraleja Madrid
Hospital Universitario La Moraleja Madrid

Autores:
La paradoja de ver como cae desfallecido, con la mirada pérdida, un joven atleta lleno de vida conmociona a sus compañeros, familiares y a toda la sociedad. Está tragedia puede ser incluso mayor si posteriormente otro familiar suyo sufre también una muerte súbita. El determinar qué casos son debidos a una mutación en un gen y por tanto que la muerte súbita sea susceptible de repetirse en otros miembros de la familia es de enorme utilidad en la prevención de estos fallecimientos.
Muchas de las etiologías principales de la muerte súbita cardiaca en los jóvenes son congénitas y / o genéticas por lo que la muerte súbita de una persona joven debe estudiarse dentro del contexto de la familia.
Durante los últimos años, se ha producido un avance muy importante en la capacidad de analizar los genes gracias a una nueva tecnología llamada secuenciación de nueva generación (NGS). Esta tecnología permite analizar simultáneamente y a un coste mucho menor que con las técnicas antiguas varios genes que podemos sospechar implicados en la muerte súbita.
Estos avances en la genética cardiovascular han añadido una visión molecular a estas enfermedades pero también han aumentado la complejidad y ambigüedad en la interpretación de las consecuencias clínicas de las anomalías genéticas en las mismas.
Tanto las pruebas genéticas premortem como post mortem permiten una evaluación del riesgo y una terapia específica para los familiares supervivientes de la víctima de una muerte súbita. El consejo, o asesoramiento genético, es un proceso de comunicación con el paciente, en el que se le explica las bases hereditarias así como las causas y consecuencias de una enfermedad hereditaria, incluyendo la implicación para su familia.
Para ello se interpreta la historia familiar y médica del paciente, se determina las pruebas genéticas más idóneas en la familia y se establece una y plan de prevención para el paciente y los miembros de su familia.
El consejo, o asesoramiento genético, ayuda a los pacientes a tomar decisiones informadas médicas y personales.
La incorporación del asesoramiento genético en la evaluación y el tratamiento de pacientes con afecciones cardíacas hereditarias, incluidas las arritmias y las miocardiopatías, se recomienda en las guías de detección precoz y seguimiento de éstas patologías y los beneficios entre otros son:
a) Ahorro de tiempo para el cardiólogo, solicitando y revisando los documentos clínicos adecuados, como informes de autopsias, pruebas genéticas realizadas etc.
b) Selección idónea del estudio genético a realizar, aquel con mayor utilidad clínica, validez analítica y nivel de detección, sí como selección del miembro
adecuado de la familia para iniciar el estudio genético (caso índice).
c) Conocimiento continuo de las actualizaciones y diversas opciones de estudios genéticos, interpretación de variantes genéticas, correlaciones entre genotipo y fenotipo.
d) Comunicación con el paciente y su familia, interpretando correctamente los resultados de los estudios genéticos y su significación clínica en el contexto de la historia del paciente y su familia, explicación de las posibles opciones reproductivas para evitar tener un hijo afecto y promover la toma de decisiones informada por parte del paciente.
Sin embargo, en la población más joven, de menos de 35 años de edad, las miocardiopatías y las arritmias hereditarias, canalopatías, tienen mucha más prevalencia.
Según datos epidemiológicos, aproximadamente:
Las enfermedades genéticas estructurales incluyen:
Las enfermedades genéticas arritmogénicas no estructurales son las enfermedades de los canales iónicos, como:
Estas enfermedades cardiacas hereditarias tienen una serie de características en común:
Por tanto, para poder clasificar con mayor rigor la variante genética encontrada y obtener los resultados informativos más concluyentes y relacionarlos con una patología, es mejor iniciar el estudio genético familiar en una persona claramente afectada.
Además, dado que a veces hay múltiples variantes genéticas que contribuyen a la enfermedad en una sola familia, lo ideal sería que la prueba se iniciara en la persona con más probabilidades de albergar todas las variantes causantes de la enfermedad y frecuentemente será el individuo de la familia con la enfermedad más grave y / o el inicio más temprano de la enfermedad.
La explicación del defecto genético, sus consecuencias sobre la fisiología cardiaca, su mecanismo de transmisión y el pronóstico que conlleva tienen una gran importancia en el momento del diagnóstico para poder aminorar las consecuencias de la enfermedad en el paciente y en sus familiares.
A aquellos pacientes con un diagnóstico antiguo de cardiopatía hereditaria se les debe ofrecer el diagnóstico genético antes de una gestación, pues es fundamental el conocer si es posible detectar la patología molecular presente en la familia para poder hacer un diagnóstico preimplantacional.
La mayoría de las enfermedades cardiacas familiares se heredan siguiendo un patrón autosómico dominante, lo que implica que un individuo portador de una
mutación tiene un 50% de riesgo de tener un hijo afecto.
Actualmente si se conoce la patología molecular presente en la familia, es posible evitar transmitir la enfermedad a los descendientes utilizando técnicas de reproducción asistida. Mediante un proceso de fertilización in vitro, es posible seleccionar aquellos embriones libres de la mutación que serán los que se transfieren a la madre evitando por tanto tener un hijo portador de la enfermedad.
El avance tecnológico de los estudios genéticos aumenta la capacidad diagnóstica por lo que a pacientes con diagnóstico clínico y estudio genético antiguo, en el que no se detectó la patología molecular, se les puede ofrecer reevaluación si cumplen los criterios diagnósticos. En estos casos podemos ofrecer la realización de un exoma completo (22.000 genes) o la secuenciación del genoma completo que ha demostrado que aumenta hasta en un 20% la posibilidad de hallar una variante patogénica.
Además, conviene revisar la historia familiar y los estudios genéticos realizados pues la segregación de una variante de significado incierto puede hacer que se reclasifique la misma como benigna o patogénica.
Desde la aparición de la secuenciación masiva o de nueva generación (NGS) los estudios de miocardiopatías hereditarias y arritmias hereditarias se ha realizado
eligiendo un número determinado de genes en los que ya se conoce su relación con la enfermedad que queremos diagnosticar.
Tiene la ventaja de mayor profundidad de secuenciación, mayor exactitud, menor número de variantes de significado incierto (VOUS o variants of unknown significance), pero la desventaja que tienen que estar rediseñándose frecuentemente por la asociación de nuevos genes a las patologías que conocemos. Esto conlleva, que casi cada laboratorio elija de forma distinta los genes incluidos en su panel, por lo que antes de la prescripción, se ha de verificar los genes estudiados en cada caso.
Secuenciación de exoma completo (WES Whole Exome Sequencing)
Los avances tecnológicos en los equipos de secuenciación masiva y en los software de análisis están haciendo que la secuenciación del exoma completo, incluso del genoma, sea una alternativa atractiva en coste y capacidad diagnóstica para el estudio de las enfermedades cardiovasculares hereditarias.
El WES nos proporciona datos de secuencia para todos los genes conocidos y es superior en capacidad diagnóstica en aquellas familias con cuadros clínicos no claros en cuanto a su diagnóstico, además de realizar también el diagnóstico en los casos sindrómicos.
Es importante señalar, que se pueden hallar mutaciones causantes de enfermedad en genes no relacionados con la patología que se estudia, es decir hallazgos incidentales. Esta posibilidad se debe de advertir durante el asesoramiento genético previo a la realización de la prueba. Esta ampliamente establecido el consenso de estudiar en todos los casos en los que se realiza un WES 59 genes relevantes recomendados por el grupo de expertos del American College of Medical Genetics.
Secuenciación del genoma completo. (WGS Whole Genome Sequencing)
En poco tiempo será la prueba de elección, pues además de la información proporcionada por el WES nos podrá informar sobre alteraciones genéticas difíciles de detectar actualmente con los paneles o con el WES como son las delecciones y duplicaciones grandes. Incluso podrá dar información de variantes importantes en Farmacogenómica, que determinarán la dosis de fármacos adecuadas para cada individuo y sus interacciones.
Se hará realidad la medicina personalizada, el tratamiento adecuado para la persona adecuada a la dosis adecuada.
Aunque se han hecho públicas nuevas directrices que han intentado estandarizar y aumentar la rigurosidad de la interpretación, con criterios más claros y con más evidencia, las interpretaciones proporcionadas para una variante determinada pueden diferir entre los laboratorios.
Además, pueden ocurrir actualizaciones y revisiones de la interpretación del laboratorio a medida que se obtiene más información de cohortes más grandes de pacientes.
Una segunda etapa es la interpretación realizada por el clínico. Es importante destacar que los resultados de las pruebas moleculares deben interpretarse en el contexto del paciente y deben integrarse con la información sobre la presentación, el curso de la enfermedad, los antecedentes familiares y los hallazgos del diagnóstico clínico.
La información del historial familiar y la segregación de una posible variante patogénica dentro de la familia pueden ser importantes para guiar la interpretación clínica de los resultados de las pruebas genéticas, especialmente cuando se identifican nuevas variantes genéticas. Los resultados de las pruebas genéticas son de naturaleza probabilística y deben interpretarse en el contexto de los antecedentes médicos y familiares del paciente.
La colaboración y comunicación entre cardiólogos, genetistas clínicos, genetistas moleculares y patólogos es la mejor garantía de la correcta interpretación de los estudios genéticos y alcanzar la máxima utilidad clínica para el paciente.
Tanto si el resultado es que no se han hallado variantes patogénicas como si son de significado incierto o patogénicas, es esencial esta segunda sesión de asesoramiento genético. En caso de que no se hallen variantes patogénicas, dependiendo de la sospecha clínica inicial y de la historia familiar puede estar indicado el realizar estudios más completos, exoma o genoma, o bien determinar que este paciente no ha heredado la enfermedad presente en su familia.
Debido a que la tasa de detección de las pruebas genéticas para cada enfermedad cardiovascular hereditaria es inferior al 100%, un resultado negativo de la prueba genética no descarta una causa genética heredada. Un resultado negativo en un paciente, simplemente significa que las pruebas actualmente disponibles no pueden identificar la causa específica de la enfermedad en la familia estudiada.
Si el resultado es la presencia de una variante de significado incierto, es posible que se decida hacer un estudio de segregación de la variante en la familia y poder reclasificarla como benigna o patogénica. Si no es posible realizar el estudio de segregación o no está indicado, todas las guías clínicas señalan que las variantes de significado incierto no han de conducir nuestra actuación clínica y por tanto no deben de tenerse en cuenta para el manejo del paciente ni de sus familiares.
Este resultado no se puede usar para pruebas genéticas predictivas en cascada en familiares. En cambio, los familiares en riesgo deben continuar la vigilancia clínica. La familia también debe mantener una relación con el servicio de genética cardiovascular, ya que las variantes de significado incierto se pueden reclasificar con el tiempo.
Cuando las pruebas del probando identifican una variante patogénica, el estudio nos proporciona una confirmación molecular del diagnóstico clínico.
El gen específico, así como la variante específica, deben investigarse para obtener información pronóstica. Por ejemplo, en la arritmia denominada síndrome del QT largo (LQTS), el gen con la alteración patógena determina el subtipo de enfermedad (LQT1, LQT2, etc.) y puede proporcionar información sobre los desencadenantes más probables de arritmias ventriculares (p. Ej., Actividad física o emoción intensa en LQT1, estímulo auditivo o período posparto en LQT2).
La ubicación y las propiedades de la variante específica también se pueden usar, junto con parámetros clínicos adicionales, para ayudar con la estratificación de riesgo de muerte súbita.
La identificación de una variante patogénica también permite la prueba en cascada de los parientes. Aquellos familiares que dan positivo para la variante deben continuar con la vigilancia clínica, mientrasque aquellos que dan negativo para la variante probablemente no estén en riesgo y no requieran vigilancia clínica.
La prueba de un paciente sano para una variante patogénica identificada en la familia se denomina prueba genética predictiva. Esta prueba debe realizarse después de un asesoramiento genético previo, porque un resultado positivo no sólo puede tener implicaciones para la atención cardíaca del paciente, sino también puede tener un impacto psicosocial en el individuo y sus descendientes.
En la sesión de asesoramiento o consejo genético posterior a la prueba, la explicación de los resultados, tanto desde el punto de vista técnico como médico, puede ser difícil de entender por muchas familias. Es muy importante que el paciente y sus familiares comprendan las implicaciones del resultado para todos los miembros de la familia. En esta sesión es posible que el paciente si entienda lo que se le explica pero que posteriormente no pueda transmitirlo a sus familiares o a sus médicos.
Por esto es esencial que además del informe con los resultados del laboratorio, se entregue al paciente un informe de consejo o de asesoramiento genético, en el que se haga un esfuerzo en explicar de forma lo más sencilla posible la consecuencia de los resultados para el paciente y su familia. En ocasiones es necesario también entregar una carta para los familiares del paciente en el que se recomiende que acudan a un servicio de genética, para determinar si están en riesgo de padecer la enfermedad estudiada en su pariente.
Por último, es muy importante no olvidar en esta sesión, si viene al caso, explicar al portador de una variante patogénica el riesgo de transmisión a sus descendientes y las opciones reproductivas que tenemos actualmente para que esto no ocurra. La selección de embriones libres de la mutación mediante diagnóstico preimplantacional antes de la transferencia en un ciclo de fertilización in vitro, permite el tener hijos no portadores de la mutación y que no padecerán la enfermedad en el futuro.
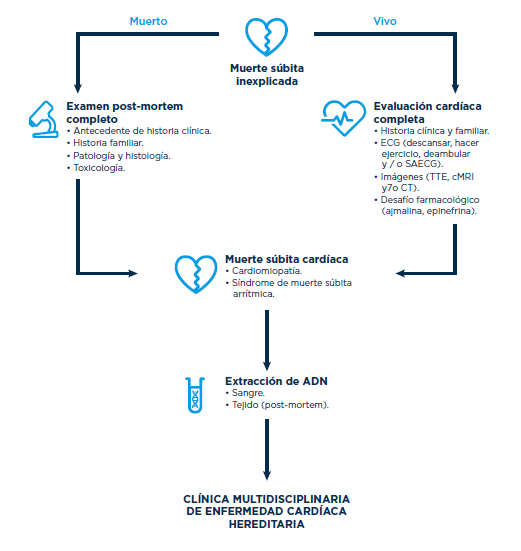
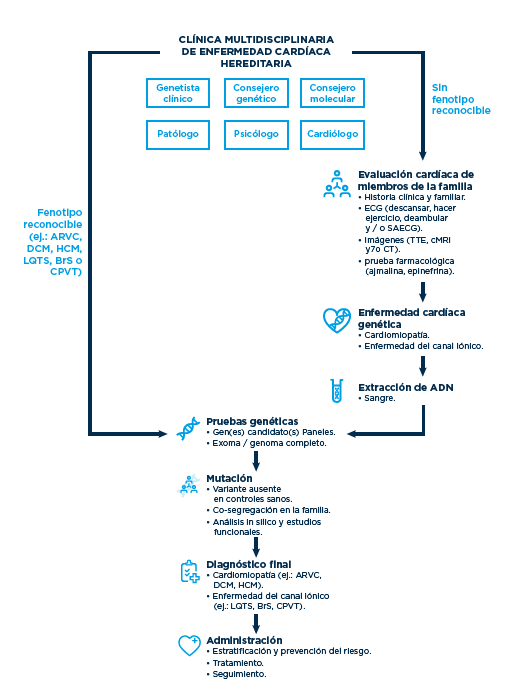
Explicación del cuadro
En la figura adjunta, modificada de A.S. Amin, A.A.M. Wilde/ Progress in Pediatric Cardiology 45 (2017) 49-54 se propone un algoritmo de actuación en caso de muerte súbita dentro de una unidad integrada de atención a las enfermedades cardiovasculares hereditarias.
Conclusiones
En su conjunto las enfermedades cardiovasculares hereditarias monogénicas afectan a un segmento importante de la población. El avance en la tecnología de los estudios genéticos ha aumentado la capacidad diagnóstica en éstas enfermedades detectando de forma más eficiente personas sanas en las cuales se desarrollarán una de ellas permitiendo el establecimiento de medidas preventivas para aminorar las consecuencias de su padecimiento, en especial de la muerte súbita cardiaca así como la trasmisión a sus descendientes. En la aplicación clínica práctica y sobretodo en la prevención los profesionales en genética médica y asesoramiento o consejo genético juegan un papel fundamental junto con los cardiólogos en el manejo de los pacientes y familias afectas. La interpretación del resultado del estudio genético en el contexto de la historia clínica del paciente es fundamental para poder prevenir las consecuencias de ser portador de una mutación en uno de estos genes como es la muerte súbita cardiaca.
Sanitas Hospitales ha ampliado su oferta de Cirugía Cardiaca en Madrid con el establecimiento de una red integrada que está formada por un mismo equipo ...
El Hospital La Moraleja se mantiene como 6º hospital privado con mejor reputación en el Monitor de Reputación Sanitaria 2023








